理論創薬研究所の金子信人です。
今回は化学反応の遷移状態を予測するオープンソースソフトウェアを紹介します。
参考文献
Locating Transition States by Variational Reaction Path Optimization with an Energy-Derivative-Free Objective Function
(Shin-ichi Koda and Shinji Saito, Journal of Chemical Theory and Computation, 2024, 20, 7, 2798–2811.)
https://doi.org/10.1021/acs.jctc.3c01246
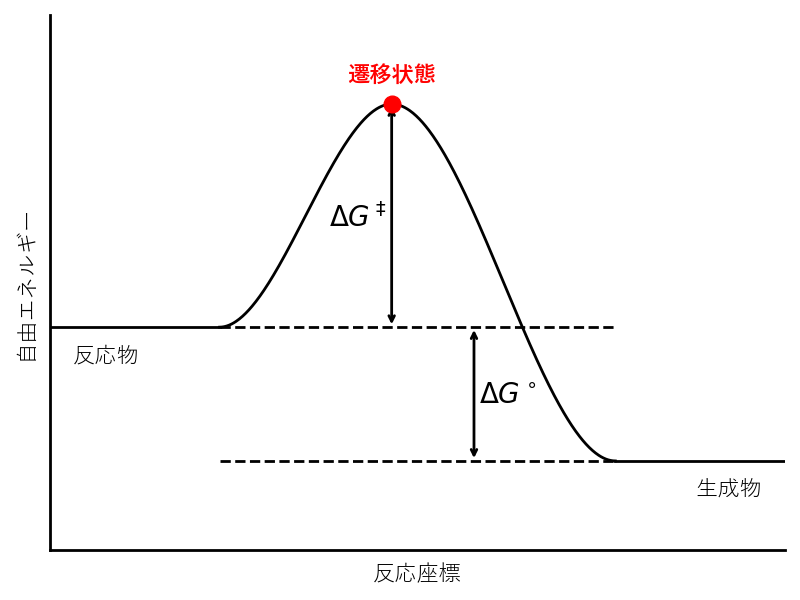
化学反応において反応物が生成物へと変化する際にはエネルギーの高い状態を経由します。この極大値を遷移状態といい、反応物とのエネルギー差ΔG‡を活性化エネルギーといいます。
遷移状態はその反応が進行するかどうか、あるいは複数の反応経路が考えられる場合にどれが確からしいかを評価する上で非常に重要となってきます。一方でこの遷移状態はエネルギー的には不安定な状態であるため実験的に検出することは困難であり、そのため今日では計算科学的手法が広く用いられています。
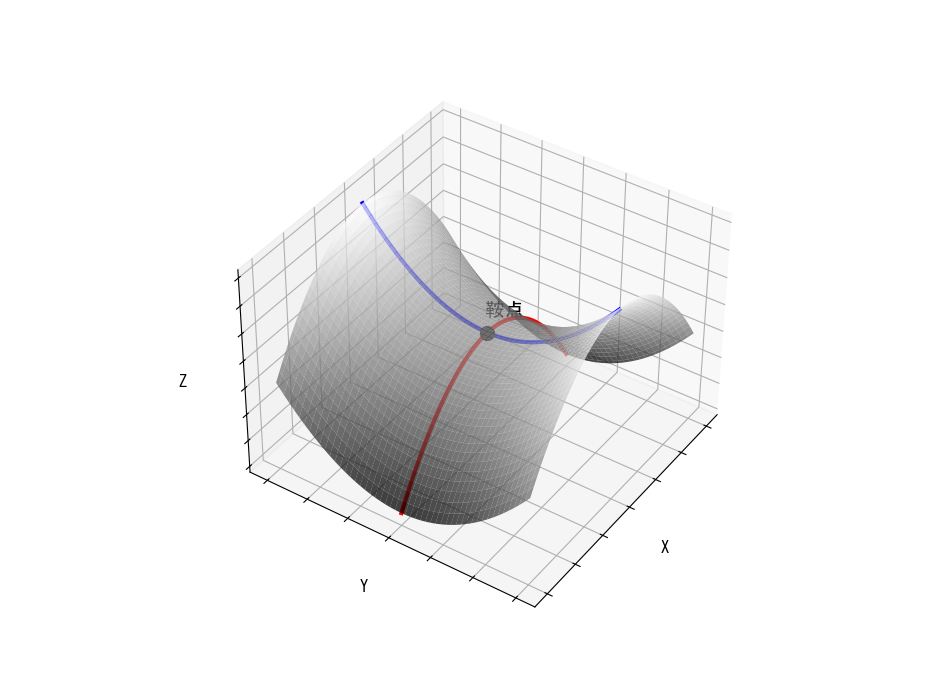
三次元のエネルギーダイアグラム上で遷移状態は下図に示す鞍点(suddle point)に該当し、ある方向(青線)からは極小値、別方向(赤線)からは極大値を取るような点となっており、最小エネルギーの算出とは異なる計算方法が求められます。
遷移状態の予測にはいくつかの方法が開発されており、MEP(Minimum Energy Path)法は初期構造を反応が進行する方向へと変化させたときの近傍の最小エネルギー経路を求める計算方法で、NEB(Nudged Elastic Band)法は各原子をバネで結んだときの伸び縮みからエネルギーを計算する方法です。しかしながら前者は遷移状態に近い初期構造を用意しなければならない点、後者は計算コストが非常に高い点が問題となっていました。
今回紹介するDirect MaxFlux法では少数の初期座標から効率的に計算することで、計算コストを低減しつつ高精度な遷移状態予測を可能にしたものになります。
動作環境
Intel Core i7-1265U
Windows 11 Pro
conda 25.3.1
Direct MaxFlux methodのインストール
Pythonの仮想環境を作成し、必要なライブラリのインストールを行い、続けてGitからDirect MaxFlux method (dmf)をインストールします。
$ conda create -n dmf python=3.12 -y
$ conda activate dmf
$ conda install numpy==1.26 scipy ase cyipopt jupyter -y
$ pip install git+https://github.com/shin1koda/dmf.git本環境ではnumpy-1.26.4 scipy-1.15.2 ase-3.25.0 cyipopt-1.5.0 direct_maxflux-0.1.0がインストールされました。
Direct MaxFlux methodによる遷移状態の予測
Githubで配布されているサンプルスクリプトを実行してみましょう。
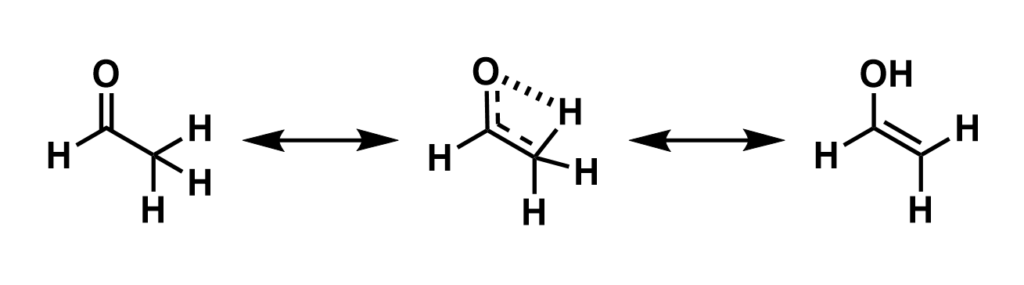
ダウンロードしたファイルのうち、react.xyzが反応原料、prod.xyzが生成物の構造ファイルとなっており、アセトアルデヒドのエノールへの互変異性反応となっています。
$ git clone https://github.com/shin1koda/dmf.git
$ cd dmf
$ python sample.py本環境では数秒で計算が実行され、最終行にOptimal Solution Foundと表示されれば遷移状態が計算されています。いくつかのファイルが出力されますが、そのうちsample_fin.trajが最終的な遷移状態構造の含まれるものになります。
windowsではトラジェクトリファイルが正しく開けないことがあるのでjupyter notebookで表示してみましょう。jupyter notebookを起動し、以下のスクリプトを実行します。
from ase.io import read
import nglview as nv
from IPython.display import display
from ase.io import write
atoms_list = read("sample_fin.traj", index=":")
for i, atoms in enumerate(atoms_list):
print(f"structure {i}")
view = nv.show_ase(atoms)
view.add_representation("ball+stick")
display(view)5つの構造が描画され、そのうちstructure0はreact.xyzの構造、structure4はprod.xyzの構造であり、structure1-3のいずれかが遷移状態となります。今回はstructure1が最も遷移状態に近い構造となっています。
より実用的な例としてブタジエンと無水マレイン酸のディールズアルダー反応の遷移状態を求めてみましょう。
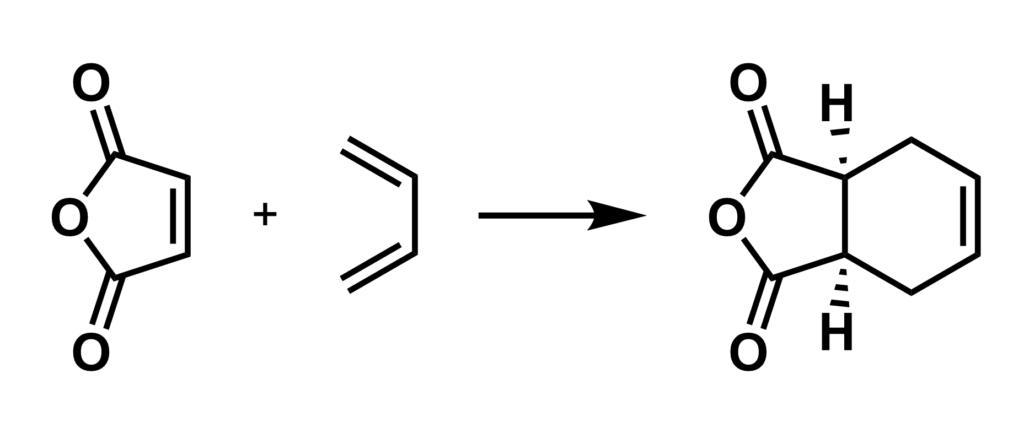
反応物・生成物それぞれの構造ファイルをchemdraw等を使って作成します。
このとき反応物・生成物それぞれの原子のラベル番号が対応するように注意します。
19
react
C 0.41460 0.70090 1.79300
C 0.51180 -0.67850 2.20430
C -0.55420 1.54310 2.17170
C -0.50720 -1.54590 2.20840
H 1.23020 1.06960 1.17300
H 1.50700 -1.02370 2.47880
H -0.54110 2.57740 1.84350
H -1.36190 1.23500 2.82700
H -0.34980 -2.57740 2.50780
H -1.50700 -1.25880 1.90520
C -2.35400 0.69050 -0.02110
C -2.33320 -0.63580 -0.00490
C -3.74810 1.10390 -0.00020
O -4.51230 -0.00630 0.02710
C -3.71380 -1.09240 0.02560
O -4.06290 -2.25680 0.04600
O -4.13400 2.25680 -0.00610
H -1.52790 1.37880 -0.04600
H -1.48770 -1.29930 -0.0128019
prod
C 1.57650 0.36120 0.63560
C 1.52190 -0.83050 0.03150
C 0.43280 1.32030 0.68720
C 0.30920 -1.35890 -0.66600
H 2.49810 0.66410 1.12990
H 2.40420 -1.46870 0.03310
H 0.79580 2.31390 0.39530
H 0.12260 1.41460 1.73610
H 0.54890 -1.40460 -1.73610
H 0.13930 -2.39260 -0.34300
C -0.74710 0.95280 -0.21770
C -0.95310 -0.52510 -0.48700
C -2.01000 1.27970 0.51550
O -2.49250 0.16320 1.11560
C -1.80350 -0.92420 0.68210
O -1.94090 -2.04950 1.13030
O -2.49810 2.39260 0.62320
H -0.70570 1.50160 -1.16490
H -1.59560 -0.66160 -1.36860反応物をreact.xyz、生成物をprod.xyzとして、sample.pyと同じフォルダに保存し、sample.pyを実行します。
先ほどと同様にjupyter notebookで生成された構造を表示してみましょう。
今回はstructure2が最も遷移状態に近い構造として表示されました。
次に得られた構造からエネルギーを計算して、ダイアグラムを作成してみましょう。
jupyter notebookからそれぞれの構造を書き出します。
write('molecule0.xyz', atoms_list[0])
write('molecule1.xyz', atoms_list[1])
write('molecule2.xyz', atoms_list[2])
write('molecule3.xyz', atoms_list[3])
write('molecule4.xyz', atoms_list[4])エネルギーの計算にはASEデフォルトのEMP(effective-medium theory)では精度が低いので半経験的計算法のxTB(Extended Tight Binding)を使用します。
$ conda install xtb xtb-python以下のプログラムでエネルギー計算とプロットをしてみましょう。
import matplotlib.pyplot as plt
from ase.io import read
import numpy as np
from xtb.ase.calculator import XTB
import sys
import os
NUM_INTERMEDIATES = 5
EV_TO_KCAL_PER_MOL = 23.0605
def plot_from_xyz_sequence():
filenames = [f'molecule{i}.xyz' for i in range(NUM_INTERMEDIATES)]
calculator = XTB()
# --- 1. 各ファイルのエネルギーを計算 ---
energies_ev = []
labels = []
for filename in filenames:
atoms = read(filename)
label = os.path.splitext(os.path.basename(filename))[0]
labels.append(label)
atoms.set_calculator(calculator)
energy = atoms.get_potential_energy()
energies_ev.append(energy)
print(f"{filename}: {energy:.6f} eV")
print("--- 計算完了 ---\n")
# --- 2. 相対エネルギーの計算 (全体の最小エネルギーを0とする) ---
energies_ev = np.array(energies_ev)
min_energy_ev = energies_ev.min()
relative_energies_ev = energies_ev - min_energy_ev
# eVからkcal/molへ単位変換
relative_energies_kcal = relative_energies_ev * EV_TO_KCAL_PER_MOL
print("--- 相対エネルギー (単位: kcal/mol) ---")
for i, rel_e in enumerate(relative_energies_kcal):
print(f" {labels[i]:<15s}: {rel_e:+.4f} kcal/mol")
print("---------------------------------------")
# --- 3. エネルギーダイアグラムのプロット ---
plt.style.use('seaborn-v0_8-whitegrid')
fig, ax = plt.subplots(figsize=(12, 7))
x_values = range(len(relative_energies_kcal))
ax.plot(x_values, relative_energies_kcal, marker='o', linestyle='-', color='dodgerblue', zorder=2)
ax.set_title('Reaction Energy Profile', fontsize=16)
ax.set_xlabel('Structure', fontsize=12)
ax.set_ylabel('Relative Energy (kcal/mol)', fontsize=12)
ax.set_xticks(x_values)
ax.set_xticklabels(labels, rotation=45, ha="right")
ax.tick_params(axis='both', which='major', labelsize=10)
fig.tight_layout()
plt.show()
if __name__ == '__main__':
plot_from_xyz_sequence()計算が実行されるとエネルギーとダイアグラムが表示されます。
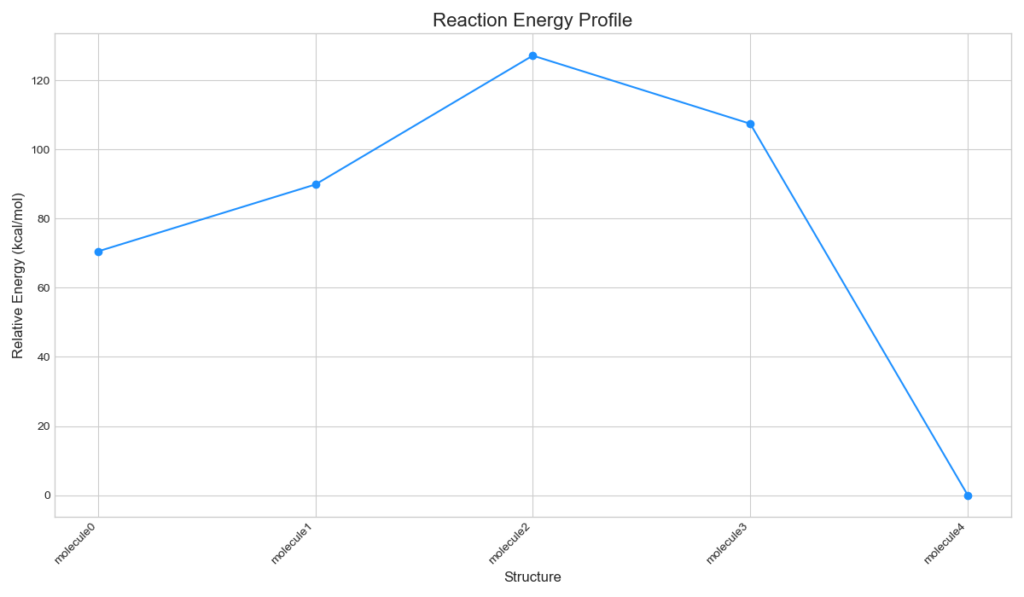
遷移状態に近いstructure2が最もエネルギーが高い状態として計算されました。
おわりに
従来、遷移状態の計算には初期構造の作り方や計算手法の選定など様々なノウハウが必要とされてきました。このように単純なインプットのみで計算ができるようになると合成化学者にとっても計算化学がより身近なものになってくると思います。
また、今回紹介したプログラムはASE(Atomic Simulation Environment)を用いて計算を行っています。そのため外部ソフトウェアを使ってより精度の高い計算を実行することも可能です。そのため必要に応じてDFT計算や虚振動解析、IRC計算へと移行していくことができます。
前回記事
GESimによる分子類似性評価
https://www.insilico.jp/blog/2025/06/19/gesim_similarity/
次回記事
Chempropを活用した機械学習による化学特性予測
https://www.insilico.jp/blog/2025/09/11/chemprop/
Category: AI創薬関連