前回から大分時間が経ってしまいましたが、今回も部分構造検索の応用の話題です。ドッキング計算結果のSDファイルから必要な条件を満たすドッキングポーズを選出するプログラムを紹介します。
ドッキングポーズ多すぎ問題
一般的なドッキング計算では化合物がタンパク質に対して色々なポーズで結合した結果が得られます。様々なドッキングシミュレーション用のプログラムがありますが、デフォルトの計算の設定だとスコアの良いものから数個〜10個程度のポーズが出力されます。とにかくドッキングポーズがいっぱい出てきて、その一つ一つがもっともらしく見えるから困るのです。そのような結果から正しいと思われるもの選択する必要があるのですが、単純にスコアの最も良いものを選択するだけでなく、化合物のファーマコフォアの結合様式も考慮に入れるとより正しいドッキングポーズを選択できる場合があります。他にもMDシミュレーションを利用する方法も考えられますが、大量の化合物ライブラリーをスクリーニングする場合には計算コストが大きすぎて使えないでしょう。
ここで紹介するプログラムはドッキング計算結果(SDファイル)とタンパク質(PDBファイル)を入力して、化合物のある部分構造が設定された条件を満たすもののみを出力するものです。
問題設定
HDAC4へのドッキング計算を例題として扱います。HDAC(ヒストン脱アセチル化酵素)は活性中心に亜鉛イオンを持っており、その阻害剤は亜鉛イオンに配位して結合することが必須であると考えられています。亜鉛に配位結合する部分構造として有名なものの1つがヒドロキサム酸です。HDAC阻害剤と言えばヒロドキサム酸です。下の図はPDBID:4CBTの阻害剤結合ポケット周りの図です。
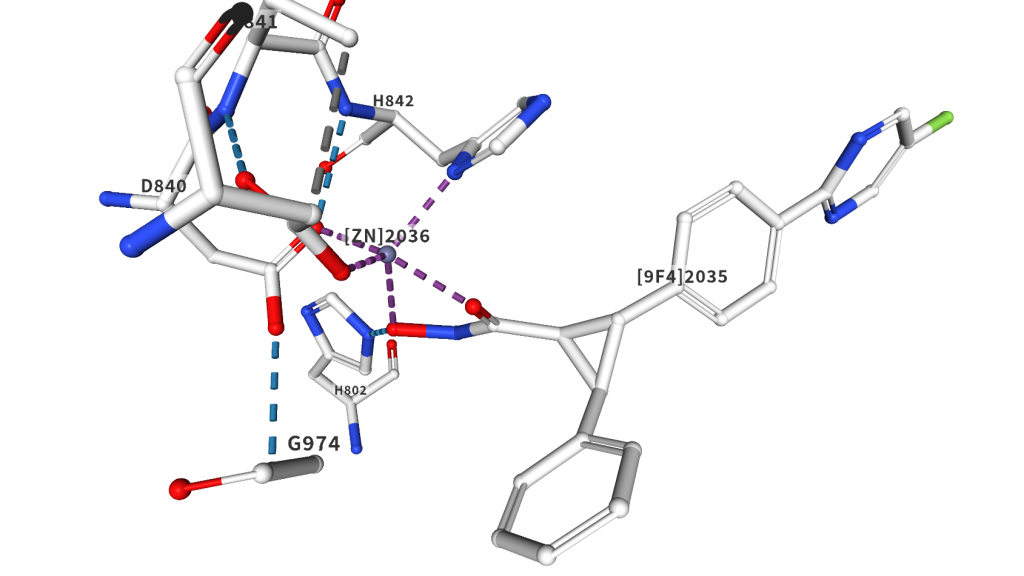
阻害剤のヒドロキサム酸がガッツリ結合していることが見て取れます。この結晶構造の場合は亜鉛とヒドロキサム酸の中心との距離は2.2Å程度です。
ドッキング計算のプログラムは”smina“を使いました。これはAutoDock Vinaからforkしたオープンソースプロジェクトで開発されています。Vinaと比べてSDファイルを入力できるなど非常に使い勝手が向上しております。そしてドッキングする化合物は共結晶構造(PDBID:4CBT)のものを利用します。所謂再ドッキングと言うやつです。どのようなドッキングソフトでも、通常は1化合物あたり10個程度のポーズが出力されます。今回紹介するプログラムは、複数出力されるドッキングポーズからヒドロキサム酸が亜鉛にキレートしているようなポーズを抽出するものです。抽出条件としては亜鉛とヒドロキサム酸の中心との距離が3.5Å以内であるとします。
ドッキング結果
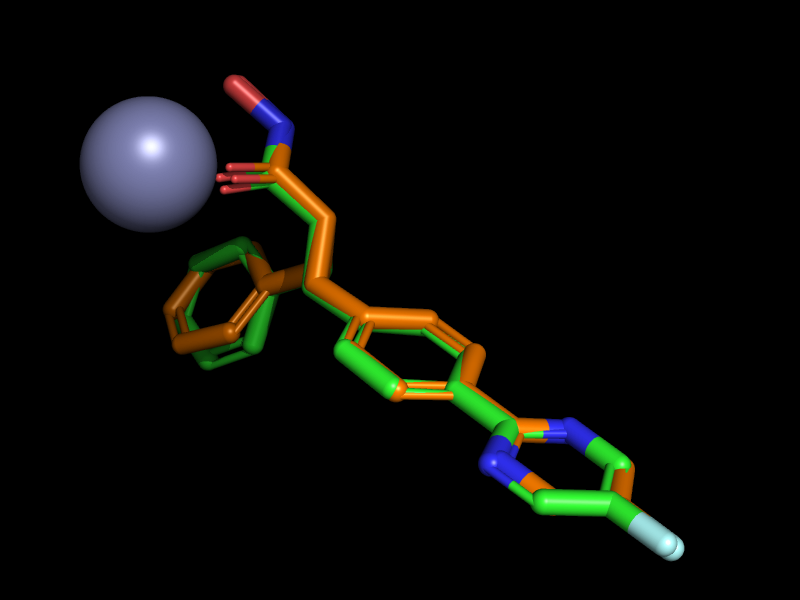
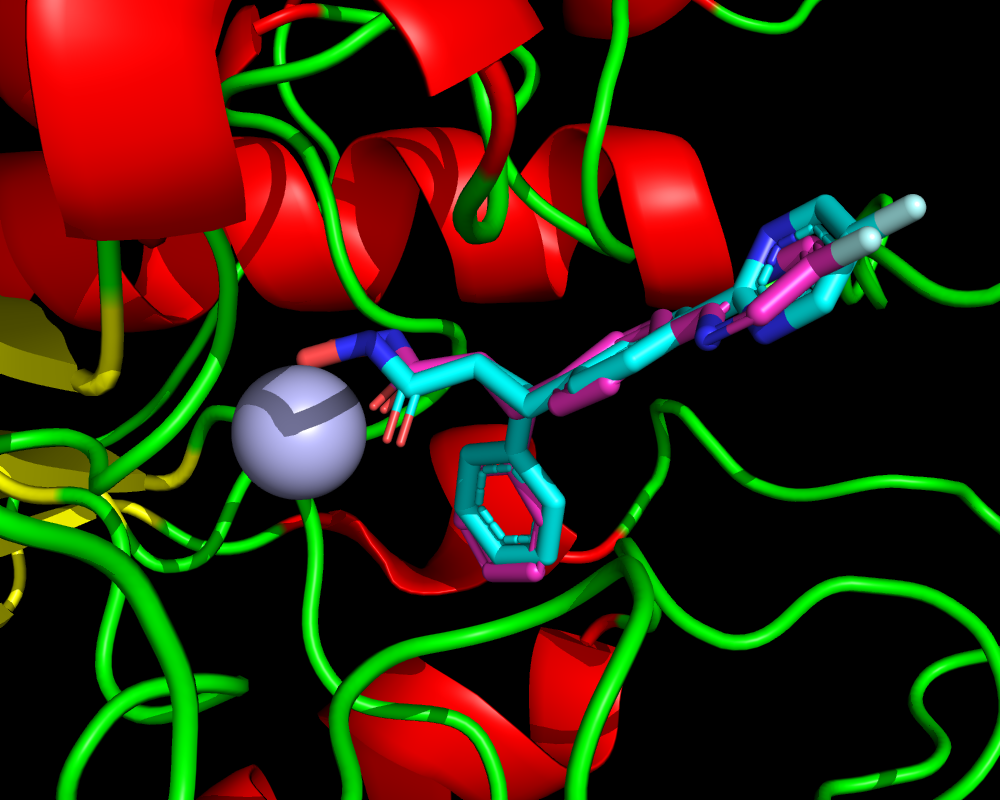
ここではドッキング計算の詳細の解説は省いて結果ファイルだけ示します。ファイルはSD形式で”smina_4cbt.sdf“です。ドッキングスコアの良い順にソートされています。このドッキング結果をPyMOLなどで4CBTの構造と比較すると、下図のように、1番目と2番目が亜鉛と相互作用するようなドッキングポーズであることが分かります。緑色が1番目のポーズで橙色が2番目のポーズです。その他の8個のポーズは省略しています。したがって今回紹介するプログラムを使うと以下のような2ポーズのみをSDファイルとして抽出できるわけです。
プログラム解説
プログラムは”HAselection.cpp“です。以下にソースコードの一部を抜粋します。ポイントとしては49、50行目で部分構造検索するところで、45行目で定義されたSMARTSパターン(ヒドロキサム酸)にマッチした部分構造の原子インデックス番号を取得しています。このインデックス番号により、続く51〜62行目でヒドロキサム酸の中心座標を計算しています。そして64〜66行目で亜鉛原子との距離を計算して、68行目で3.5Å以内かどうか判定しています。
35 OBMol prot;
36 conv1.Read(&prot, &ifs_prot);
37 OBAtom* zinc;
38 for (auto ii = prot.BeginResidues(); ii != prot.EndResidues(); ii++) {
39 if ((*ii)->GetNum() == 2036) {
40 zinc = *((*ii)->BeginAtoms());
41 }
42 }
43
44 OBSmartsPattern smp;
45 smp.Init("[OX1]NC(=O)[#6]");
46
47 OBMol mol;
48 while (conv2.Read(&mol, &ifs_sdf)) {
49 smp.Match(mol);
50 vector<vector<int> > mlist = smp.GetUMapList();
51 double gx = 0;
52 double gy = 0;
53 double gz = 0;
54 for (int i = 0; i < mlist[0].size() - 1; i++) {
55 OBAtom* a = mol.GetAtom(mlist[0][i]);
56 gx += a->x();
57 gy += a->y();
58 gz += a->z();
59 }
60 gx /= (double)(mlist[0].size() - 1);
61 gy /= (double)(mlist[0].size() - 1);
62 gz /= (double)(mlist[0].size() - 1);
63
64 double d = (zinc->x() - gx) * (zinc->x() - gx) + (zinc->y() - gy) * (zinc->y() - gy) +
65 (zinc->z() - gz) * (zinc->z() - gz);
66 d = sqrt(d);
67
68 if (d < 3.5) {
69 conv2.Write(&mol, &ofs);
70 }
71 }
72 }このプログラムのコンパイルと使い方は以下の通りです。a.outの第一引数に参照するPDB構造を指定します。第二引数にはドッキング結果のSDファイルを指定します。第三引数は出力するSDファイル名を指定します。”out.sdf”中のドッキングポーズの数が2個であれば正しく計算されているかと思います。
$ g++ -std=c++11 HAselection.cpp -I/home/username/app/include/openbabel-2.0 -L/home/username/app/lib -lopenbabel
$ ./a.out 4cbt.pdb smina_4cbt.sdf out.sdf今回のプログラムは単純に亜鉛原子とヒドロキサム酸の中心との距離を計算していますが、単に近いかどうかしか見ておりません。できればヒドロキサム酸の向きも考慮すべきです。その様なプログラムの書き方(アルゴリズム)は色々有るかと思います。私が思いつく方法の一つとしては、亜鉛原子とヒドロキサム酸中心を結ぶベクトルをaとしたとき、ヒドロキサム酸中のカルボニルの方向ベクトルbとNOの方向ベクトルcの和がベクトルaと並行かどうかを計算することです。余力が有る人はチャレンジしてみて下さい。
結局
プログラム解説までして色々しましたが、結局一番スコアの良いドッキングポーズが結晶構造に最も近かったという結果でした(^_^;)。さすがsminaと言うべきでしょうか。実は最後まで黙ってましたが、ドッキングツールによっては今回のような拘束条件を入れられるものもあるかと思います。(sminaでは拘束条件は入れられませんが)しかしながら、拘束条件を入れたドッキング計算は恣意的な感じがして、私自身はあまり好みません。
Category: Docking, OpenBabel, プログラミング関連