学生アルバイトのKさんにLigandScoutの操作に慣れてもらうため,論文で説明されているLigand-based手法の再現をお願いしました.少し苦戦したようですが,以下そのレポートです.
今回は、LigandScoutを使ったファーマコフォアモデリングに関する以下の文献を紹介し、実際に論文の一部をトレースしてみたいと思います。
Discriminating agonist and antagonist ligands of the nuclear receptors using 3D-pharmacophores
Nathalie Lagarde, Solenne Delahaye, Jean-François Zagury and Matthieu MontesEmail authorView ORCID ID profile
Journal of Cheminformatics20168:43 https://doi.org/10.1186/s13321-016-0154-2
© The Author(s) 2016 Received: 24 June 2016 Accepted: 17 August 2016 Published: 6 September 2016
著者らは、核内受容体(NR)リガンドの薬理学的な特徴を予測するために、LigandScoutを用いて、構造ベース(SB)アプローチ、リガンドベース(LB)アプローチ、二つを組み合わせたSBLBアプローチによってファーマコフォアを作成し、それぞれのアプローチを評価しています。
LBアプローチでは、図1に示すような独自のプロトコルを用いています。
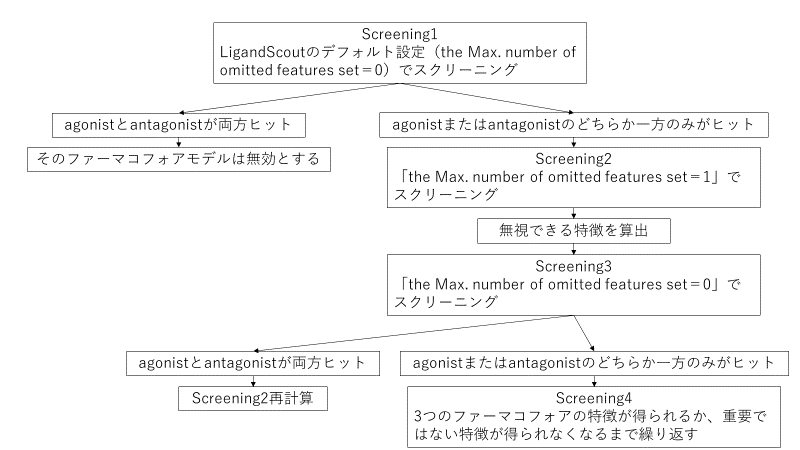
図 1 リガンドベースアプローチのプロトコル
本記事では、AR_agonistを例として、図 1のプロトコルを再現してみます。
なお、使用するデータベースである「NRLiSt BDB」は、http://nrlist.drugdesign.frからダウンロードできます。また、本文献で用いられているLigandScoutのバージョン4.0ですが、本記事ではバージョン4.4を用います。
- クラスターの作成
「Ligand-Based」ビューで「AR_agonist_ligand.ldb」(27NRs>AR>AR_ agonist>ligands)を開きます。
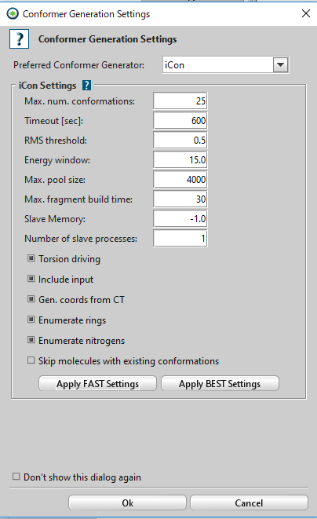
「Generate Conformations for Ligand-Set」をクリックし、下図のように設定します。ただし、著者らは「Omega-fast」を用いていますが、今回は「iCon-fast」を利用します。
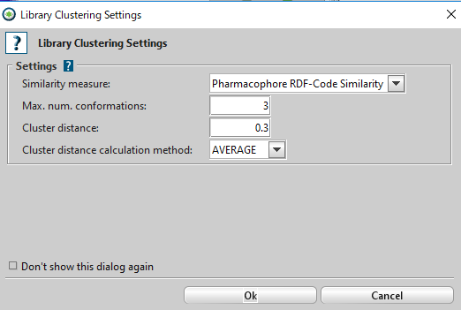
次に、「Cluster Ligand-Set」をクリックし、下図のような条件でクラスタリングを行います。
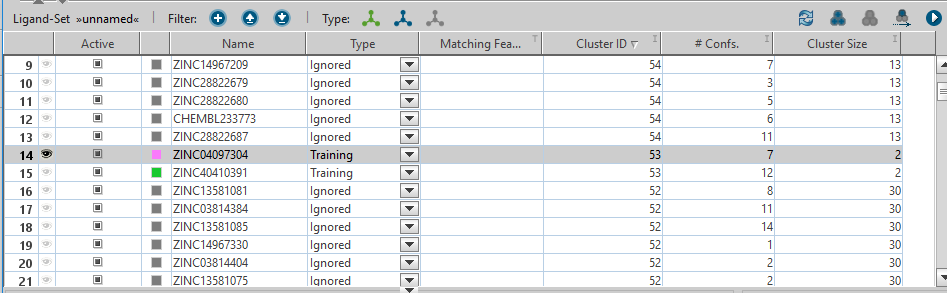
クラスタリング終了後、各構造の「Type」は「Training」となっていますので、「Set all to Ignored」をクリックし、全て「Ignored」にします。
下図のように1つの構造を選択した状態で、「Set selected Cluster IDs to Training」をクリックすると、選択した構造と同じクラスターIDを持つ構造の「Type」が全て「Training」になります。
「Create Pharmadophore」をクリックし、ファーマコフォアを作成します。
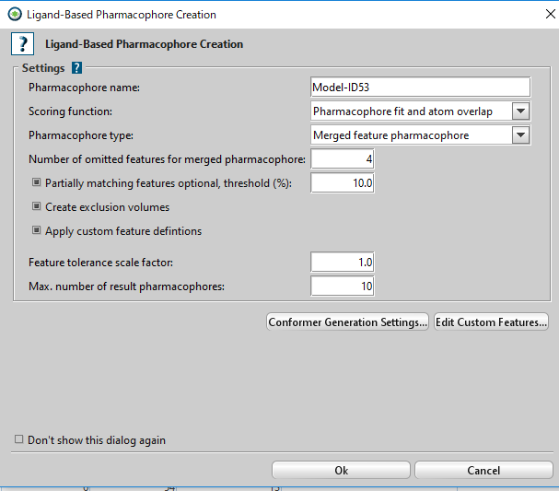
作成されたファーマコフォアを「Alignment」ビューにコピーして、保存します。
- スクリーニングの準備
NRLiSt BDBにSMILES形式で示された「AR_agonist.smi」(27NRs>AR>AR_ agonist>ligands)と「AR_agonist_decoys.smi」(27NRs>AR>AR_ agonist>decoys)を.ldb形式に変換します。今回は「Ligand-Based」ビューで「iCon-fast」を用いて行います。
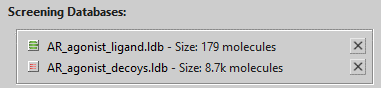
「Screening」ビューの「Screening Databases」に「AR_agonist.ldb」と「AR_agonist_decoys.ldb」をロードします。
スクリーン毎時に、下図のように「AR_agonist.ldb」が緑、「AR_agonist_decoys.ldb」が赤になっていることを確認します。
- Screening1
「File>Open」から①で作成したファーマコフォアを開きます。
LigandScoutのデフォルト設定(the Max. number of omitted features set=0)でスクリーニングを行います。
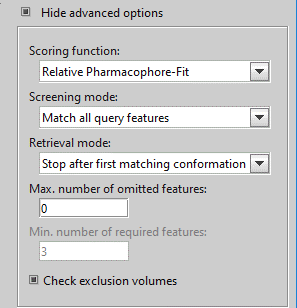
計算の結果、下図のように2個の「active」がヒットしています。全て「active」なので、操作は「Screening2」へ進みます。

- Screening2
「the Max. number of omitted features set=1」にして再度スクリーニングを行います。
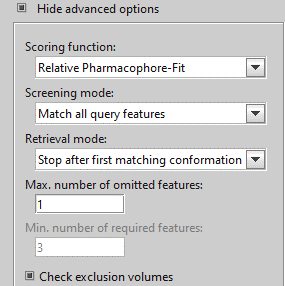
「active」と「decoy」が両方出てきます。
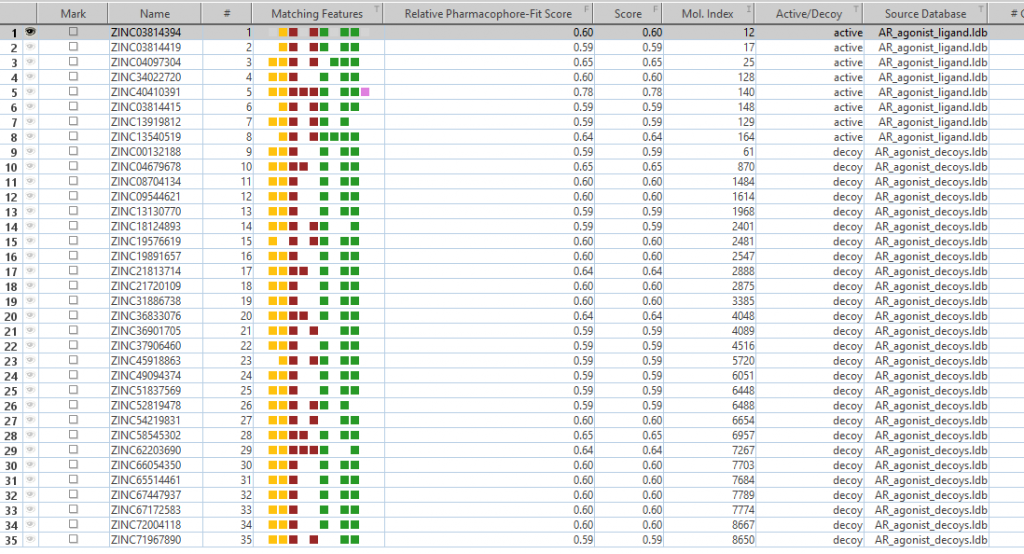
- Screening3
「Matiching Features」を見て、無視して良いと考えられる特徴を選択し、「Pharmacophore>Mark Features as Disabled」をクリックします。(下図のように選択した部分が青くなります。)適宜、排除体積を加えます。
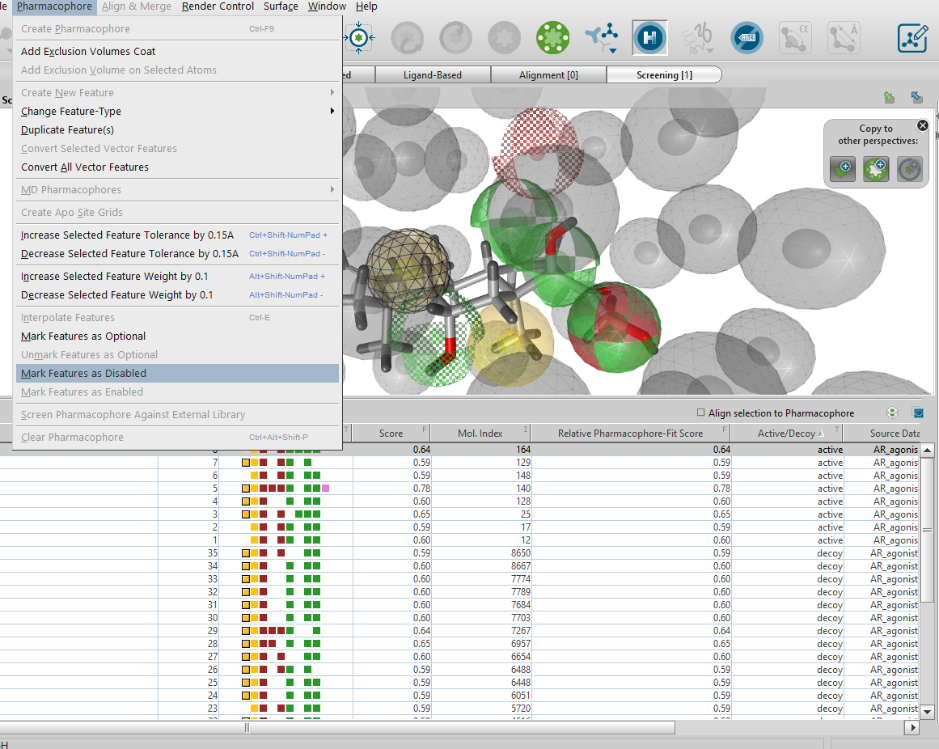
「the Max. number of omitted features set」を「0」に戻してスクリーニングを行います。上手く無視して良いと考えられる特徴を指定すると、「active」のみがヒットします。

- Screening4
3つのファーマコフォアの特徴に絞り込むか、重要ではない特徴が得られなくなるまで、スクリーニングを繰り返します。

最終的なファーマコフォアモデルを保存します。
- 最終的なヒット数の算出
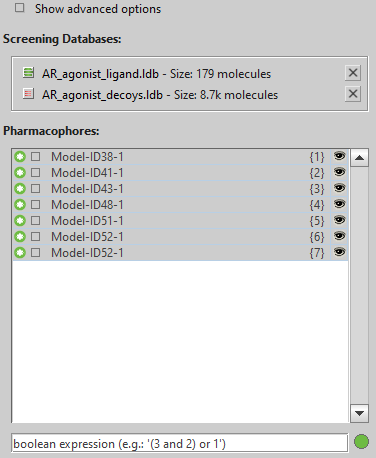
各々のクラスターIDについて作成したファーマコフォアを全て「Screening」ビューに表示し、まとめてスクリーニングを行います。「the Max. number of omitted features set」が「0」であること、「boolean expression」が「or」であることを確認してください。
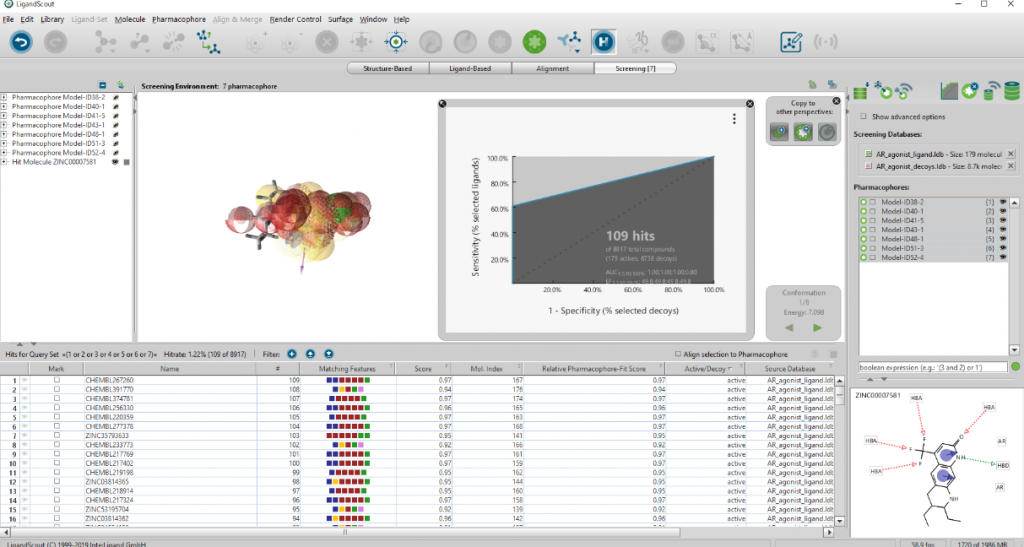
今回の計算では、「decoy」は1個もヒットせず、「active」が109個得られました。論文の結果によれば「active」が150個ほど出るはずですが、クラスタリングや無視できるとする特徴と排除体積が上手く特定できていないためだと考えられます。
以上,Kさんのレポートでした.これにStructure-basedで構築したPharmacophoreモデルを加えるとスクリーニング精度はより良くなるのではと思います.なお,このLigand-basedのプロトコルは,次図のようにKnime extensionを利用する事である程度の自動化が可能です.
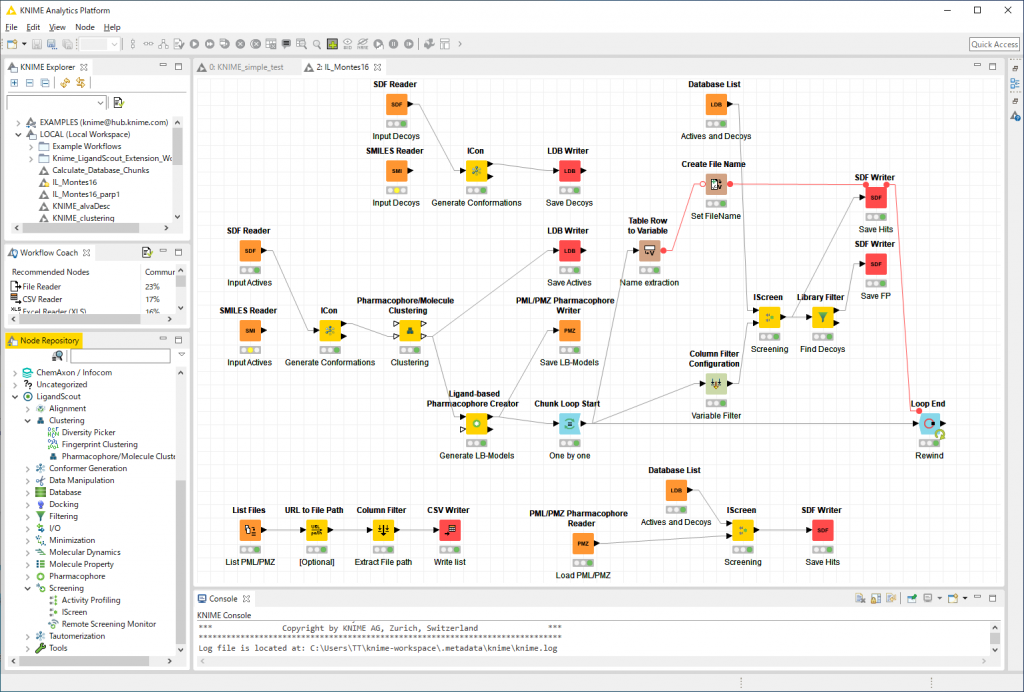
プログラミングの知識がなくても,ノードを接続していくことで繰り返し行う処理を簡略化することが出来ます.Kさんには,そのうちKnimeについても少し勉強してもらおうと考えています.
Category: LigandScout